The computer program SUPERSTRUCTURE was originally developed by
Eissner
et al. (1974), and the version used in the present work
contains improvements by
Nussbaumer & Storey (1978). A summary of the
code's main features is given by Eissner (1991).
In this approach the wavefunctions are expressed in a configuration
expansion of the type
![]()
where the basis functions ![]() are constructed from one-electron
orbitals generated in two types of potential
are constructed from one-electron
orbitals generated in two types of potential ![]() :
spectroscopic orbitals P(nl) are calculated in a
statistical Thomas-Fermi-Dirac model potential
(Eissner & Nussbaumer 1969) whereas
correlation orbitals
:
spectroscopic orbitals P(nl) are calculated in a
statistical Thomas-Fermi-Dirac model potential
(Eissner & Nussbaumer 1969) whereas
correlation orbitals ![]() are obtained in a Coulomb
potential (Nussbaumer & Storey 1978).
The scaling parameters
are obtained in a Coulomb
potential (Nussbaumer & Storey 1978).
The scaling parameters ![]() are computed variationally so as
to minimize the weighted sum of the non-relativistic energies of the terms
of the ground configuration
are computed variationally so as
to minimize the weighted sum of the non-relativistic energies of the terms
of the ground configuration
![]()
where ![]() denotes the usual statistical weight. The selection of the
configuration basis set for each sequence requires some attention.
Of course, the aim is to get level energies as close as possible to
experimental values, but it is also of primary importance to build a
well-balanced
physical model. Indeed, an excessively intricate description of a given
atomic system may introduce spurious numerical effects which can affect
considerably the calculated transition rates even when the level energies
turn out to be accurate.
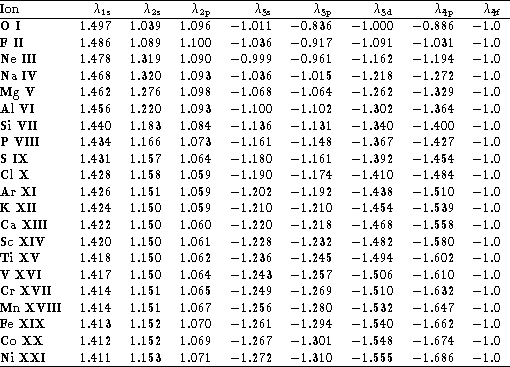
The configuration representations and scaling parameters used in this
work are listed in Tables 1-3.
denotes the usual statistical weight. The selection of the
configuration basis set for each sequence requires some attention.
Of course, the aim is to get level energies as close as possible to
experimental values, but it is also of primary importance to build a
well-balanced
physical model. Indeed, an excessively intricate description of a given
atomic system may introduce spurious numerical effects which can affect
considerably the calculated transition rates even when the level energies
turn out to be accurate.
The configuration representations and scaling parameters used in this
work are listed in Tables 1-3.
Table 1: Configuration basis sets selected for the present calculations.
Term symmetries ![]() P,
P, ![]() D and
D and ![]() S are the only ones retained in the
physical model
S are the only ones retained in the
physical model
Table 2: Scaling parameters used to generate the orbitals for the
C-like ions.
The negative scaling parameters denote correlation Coulombic orbitals
Table 3: Scaling parameters used to generate the orbitals for the
O-like ions.
The negative scaling parameters denote correlation Coulombic orbitals
In SUPERSTRUCTURE the Hamiltonian is taken to be of the form
![]()
where ![]() is the usual non-relativistic Hamiltonian and
is the usual non-relativistic Hamiltonian and
![]() is
the BP relativistic correction (Jones 1970, 1971;
Eissner et al. 1974; Eissner 1991).
Using perturbation theory, the relativistic wavefunction
is
the BP relativistic correction (Jones 1970, 1971;
Eissner et al. 1974; Eissner 1991).
Using perturbation theory, the relativistic wavefunction ![]() can
be expanded in terms of the non-relativistic functions
can
be expanded in terms of the non-relativistic functions ![]() :
:
![]()
Small fractional errors in the non-relativistic energies ![]() and
and ![]() can lead to much larger errors in the differences
can lead to much larger errors in the differences
![]() for
for ![]() . Using experimental data,
improved estimates of the non-relativistic energies can be obtained and
a modified
. Using experimental data,
improved estimates of the non-relativistic energies can be obtained and
a modified ![]() can be constructed. This semi-empirical term energy
correction (TEC) procedure was implemented in SUPERSTRUCTURE by
Zeippen et al. (1977).
In the present case, the corrections are chosen so as to move the computed
energies of the terms
can be constructed. This semi-empirical term energy
correction (TEC) procedure was implemented in SUPERSTRUCTURE by
Zeippen et al. (1977).
In the present case, the corrections are chosen so as to move the computed
energies of the terms ![]() and
and ![]() in each ion to match the
observed term separation. Previous work has shown that the
TEC procedure is efficient
and reliable when the corrections are small, i.e. when the ab initio
wavefunctions represent the system under consideration with very good
accuracy.
in each ion to match the
observed term separation. Previous work has shown that the
TEC procedure is efficient
and reliable when the corrections are small, i.e. when the ab initio
wavefunctions represent the system under consideration with very good
accuracy.
The total radiative rate for a forbidden transition is taken to be the
sum of the electric quadrupole (E2) and magnetic dipole (M1) contributions
![]()
with
![]()
and
![]()
Here ![]() is the statistical weight of the level i and energies E are
expressed in Rydbergs. From Eqs. (6 (click here)) and (7 (click here)), it is clear
that the accuracy of the calculated A-values depends
strongly on the quality of the wavefunctions used for evaluating the line
strengths S. Moreover one can see that a relatively small error in the energy
difference
is the statistical weight of the level i and energies E are
expressed in Rydbergs. From Eqs. (6 (click here)) and (7 (click here)), it is clear
that the accuracy of the calculated A-values depends
strongly on the quality of the wavefunctions used for evaluating the line
strengths S. Moreover one can see that a relatively small error in the energy
difference ![]() can mar an otherwise reliable calculation: note the
exponents 5 and 3 respectively in Eqs. (6 (click here)) and (7 (click here)).
To eliminate
such a drastic cause of inaccuracy, experimental level separations from the
extensive work of Edlén (1983, 1985) have replaced computed
energies in this work.
can mar an otherwise reliable calculation: note the
exponents 5 and 3 respectively in Eqs. (6 (click here)) and (7 (click here)).
To eliminate
such a drastic cause of inaccuracy, experimental level separations from the
extensive work of Edlén (1983, 1985) have replaced computed
energies in this work.
{kind=link}
{kind=link}
{kind=link}